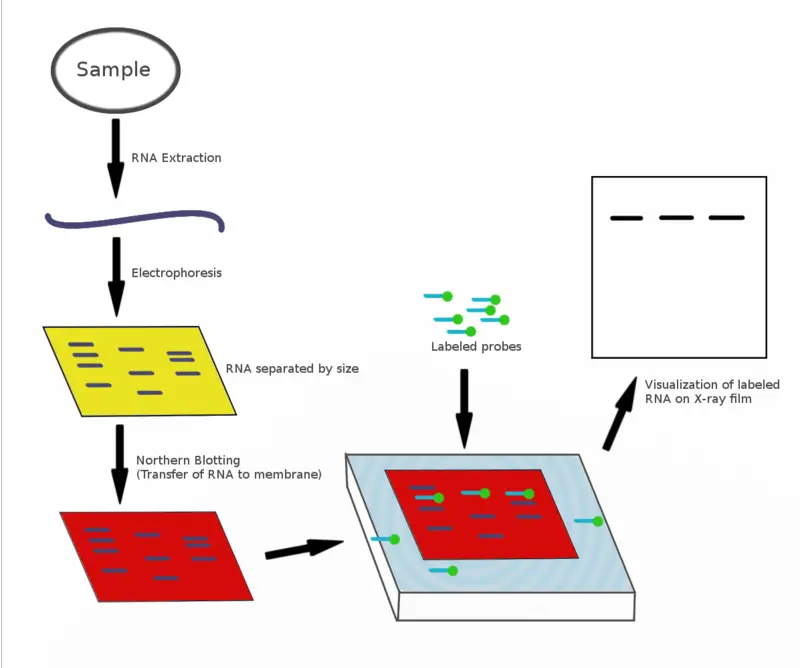
The northern blot, or RNA blot, is a technique utilized in molecular biology research to examine gene expression through the detection of RNA (or isolated mRNA) in a sample.
Using northern blotting, it is possible to observe cellular control over structure and function by determining gene expression rates during differentiation, morphogenesis, and disease.Northern blotting utilizes electrophoresis to size-separate RNA samples and a hybridization probe complementary to a portion or the entire target sequence for detection. The term ‘northern blot’ strictly refers to the capillary transfer of RNA from the electrophoresis gel to the blotting membrane. Nevertheless, the entire procedure is commonly known as northern blotting. The northern blot technique was devised in 1977 at Stanford University by James Alwine, David Kemp, and George Stark. Northern blotting is akin to the first blotting technique, the Southern blot, which was named after the biologist Edwin Southern.Northern blot analysis focuses on RNA instead of DNA.
Contents
What is Northern Blotting Technique?
Northern blotting is a molecular biology technique used to detect specific RNA sequences in a complex mixture, providing information on RNA length, sequence variations, and gene expression levels.
- The Northern blotting technique is a pivotal method in molecular biology utilized to analyze specific RNA molecules within a complex mixture. It is named after its predecessor, the Southern blotting technique, which was developed for DNA sequence analysis. Understanding the presence and characteristics of nucleic acid sequences in biological samples is crucial in molecular biology, making blotting techniques indispensable in the field.
- The basic principle of Northern blotting is akin to Southern blotting, with the key difference lying in the type of molecules being detected; Northern blotting specifically targets RNA sequences. This technique provides valuable information regarding the length of RNA sequences and the presence of any sequence variations. While primarily used for identifying RNA sequences, Northern blotting has also been employed for quantifying RNA sequences.
- Since its inception, Northern blotting has undergone several modifications to facilitate the analysis of various RNA types, including mRNAs, pre-mRNAs, and short RNAs. While Northern blotting was historically the primary method for RNA fragment analysis, more recent and cost-effective techniques such as reverse transcription-polymerase chain reaction (RT-PCR) have gradually supplanted its use.
- The Northern blot, also known as the RNA blot, is particularly valuable in molecular biology research for studying gene expression. It enables researchers to observe the control of cellular structure and function by determining gene expression rates during processes like differentiation, morphogenesis, and under abnormal or diseased conditions. The technique involves electrophoresis to separate RNA samples by size, followed by detection using a hybridization probe that is complementary to part or all of the target RNA sequence.
- While the term “Northern blot” technically refers only to the capillary transfer of RNA from the electrophoresis gel to the blotting membrane, the entire process is commonly referred to as Northern blotting. This technique was developed in 1977 by James Alwine, David Kemp, and George Stark at Stanford University, and it is named in homage to Edwin Southern, the biologist who pioneered the first blotting technique for DNA analysis.
Aim of Northern Blotting
To learn the technique of Northern Blotting for the detection of a specific RNA fragment in a sample
Northern Blotting principle
The principle of Northern blotting, like all blotting techniques, is based on the transfer of biomolecules from one membrane to another for analysis. In the context of Northern blotting, the focus is on RNA molecules.
Initially, RNA samples are separated by size using gel electrophoresis. Because RNA is single-stranded, it can form secondary structures through intermolecular base pairing. To prevent this and ensure accurate separation, the electrophoresis is performed under denaturing conditions, which disrupt secondary structures.
Once the RNA fragments are separated, they are transferred from the gel to a nylon membrane. Unlike nitrocellulose membranes, which are ineffective for RNA binding, nylon membranes ensure the RNA fragments are securely attached. The transfer process is crucial as it immobilizes the RNA fragments on the membrane, making them accessible for detection.
Detection is achieved by adding a labeled probe that is complementary to the RNA sequences on the membrane. This probe hybridizes with the target RNA sequences. The specificity of this hybridization process allows for the accurate identification of the RNA segments present. The labeled probes often contain radioactive or fluorescent tags, enabling visualization of the hybridized RNA fragments.
Northern blotting is particularly valuable for its ability to separate RNA segments based on size. This allows researchers to determine the sizes of RNA transcripts and analyze variations, providing insights into gene expression and the presence of specific RNA sequences. This technique, rooted in the fundamental principles of molecular biology, remains an essential tool for studying RNA dynamics in various biological contexts.
Material Required for Northern Blotting
Equipment
- Agarose Gel Rig: Used for the electrophoresis of RNA samples.
- Power Supply: Provides the electrical current required for gel electrophoresis.
- Microwave: Utilized for melting agarose and preparing the gel.
- Centrifuge: Separates different components of the RNA sample.
- PCR Machine: Amplifies specific DNA sequences if needed.
- Heating Block: Maintains a constant temperature for various reactions.
- Vacuum Gel Transfer System: Transfers RNA from the gel to the membrane.
- Nylon Membrane: Captures the RNA during the transfer process.
- UV Crosslinker: Fixes RNA to the nylon membrane by creating covalent bonds.
- Hybridization Oven: Ensures uniform temperature during probe hybridization.
- Hybridization Bottles: Contain the membrane and probe during hybridization.
- G-50 Sephadex Spin Column: Purifies labeled probes.
- Scintillation Vial: Holds samples for scintillation counting.
- Scintillation Counter: Measures radioactivity in labeled probes.
- Forceps: Handles membranes and other small items.
- Pipette Tips: Ensures accurate and sterile handling of liquids.
- 1.5 ml Polypropylene Tubes: Stores samples and reagents.
- Phosphor Screen: Detects radioactive signals from the hybridized membrane.
- Phosphor Screen Scanning Equipment: Visualizes and quantifies the signals.
- ImageQuant Software (Molecular Dynamics): Analyzes and interprets the data from the scanned images.
Chemicals and Reagents
- Agarose: Forms the gel matrix for electrophoresis.
- MOPS (3-(N-morpholino)propanesulfonic acid): Buffer used to maintain pH during electrophoresis.
- Sodium Acetate: Buffer component.
- Ethylenediaminetetraacetic Acid (EDTA) Disodium Salt Dihydrate: Chelates divalent cations to inhibit nucleases.
- NaOH (Sodium Hydroxide): Adjusts pH.
- HCl (Hydrochloric Acid): Adjusts pH.
- Formaldehyde: Denatures RNA for gel electrophoresis.
- Glycerol: Increases sample density for loading into wells.
- Ethidium Bromide: Stains RNA for visualization under UV light.
- Bromophenol Blue: Tracking dye for electrophoresis.
- Xylene Cyanol: Tracking dye for electrophoresis.
- Orange G: Tracking dye for electrophoresis.
- Formamide: Denatures RNA and stabilizes it.
- Millennium RNA Ladder (Ambion): RNA size standard.
- MgCl2 (Magnesium Chloride): Cofactor for enzymes.
- NTPs (ATP, CTP, GTP, UTP): Nucleotides required for RNA synthesis.
- [α-P32]-UTP: Radioactively labeled nucleotide for probe synthesis.
- Salmon Sperm DNA: Used as a blocking agent to prevent non-specific binding.
- NaCl (Sodium Chloride): Used in various buffer solutions.
- Sodium Citrate: Buffer component.
- Ficoll 400: Increases sample density.
- Polyvinylpyrrolidone (PVP): Blocking agent to reduce background.
- Bovine Serum Albumin (BSA): Stabilizes enzymes and reduces non-specific binding.
- SDS (Sodium Dodecyl Sulfate): Detergent used in buffers.
- Sodium Heparin: Anticoagulant.
- NaH2PO4 (Sodium Dihydrogen Phosphate): Buffer component.
- Na2HPO4 (Disodium Phosphate): Buffer component.
- Tris-HCl (pH 8.0): Buffer used to maintain pH.
- DTT (Dithiothreitol): Reducing agent to prevent oxidation.
- Triton X-100: Detergent used to permeabilize membranes.
- Spermidine (pH 7.0): Stabilizes nucleic acids during hybridization.
- Taq Buffer: Used with Taq polymerase for PCR.
- Enzymes:
- T7 Polymerase: Synthesizes RNA from DNA templates.
- Taq Polymerase: DNA polymerase used in PCR.
Solutions & buffers Preparation
1. MOPS Buffer
MOPS (3-(N-morpholino)propanesulfonic acid) Buffer is crucial for maintaining a stable pH during the electrophoresis of RNA samples.
- 10× MOPS Buffer Preparation:
- Components:
- MOPS: 0.2 M (41.852 g)
- Sodium acetate •3H2O: 80 mM (10.89 g)
- EDTA: 10 mM (0.372 g)
- Instructions:
- Dissolve the components in water.
- Adjust the pH to 7.0 with NaOH.
- Add water to make up to 1 liter.
- Store at room temperature, protected from light.
- Components:
2. Denaturing RNA Gel
Denaturing RNA Gel is used to separate RNA molecules by size during electrophoresis.
- Preparation:
- Components (for 150 ml):
- MOPS buffer (10×): 15 ml
- Agarose: 1.2% (w/v) (1.8 g)
- H2O: 132 ml
- Instructions:
- Microwave the mixture for 3-4 minutes to dissolve agarose.
- Cool until it can be held comfortably.
- Add 2.8 ml of formaldehyde.
- Components (for 150 ml):
3. Running Buffer
Running Buffer facilitates the movement of RNA through the gel during electrophoresis.
- Preparation:
- Components:
- MOPS buffer (10×): 100 ml
- Formaldehyde: 7% (from 37% stock, 19 ml)
- Instructions:
- Add water to make up to 1 liter.
- Components:
4. RNA Loading Buffer
RNA Loading Buffer ensures proper sample loading and visualization during electrophoresis.
- Preparation:
- Components (for 10 ml):
- MOPS buffer (10×): 1 ml
- Glycerol: 20% (2 ml)
- Formaldehyde: 6.5% (1.76 ml)
- Formamide: 50% (5 ml)
- Ethidium Bromide: 10 μg/ml (100 μg)
- Bromophenol Blue: 0.05% (w/v) (5 mg)
- Xylene Cyanol: 0.05% (w/v) (5 mg)
- Instructions:
- Add water to make up to 10 ml.
- Components (for 10 ml):
5. SSC Buffer
SSC (Saline-Sodium Citrate) Buffer is used in the hybridization and washing steps of the blotting process.
- 20× SSC Buffer Preparation:
- Components:
- NaCl: 3 M (175.3 g)
- Sodium Citrate: 300 mM (88.2 g)
- Instructions:
- Dissolve the components in water.
- Adjust the pH to 7.0 with HCl.
- Add water to make up to 1 liter.
- Components:
- 10× SSC Buffer Preparation:
- Dilute 500 ml of 20× SSC buffer with water to make 1 liter.
6. Transcription Buffer
Transcription Buffer is used during in vitro transcription reactions.
- 10× Transcription Buffer Preparation:
- Components (for 10 ml):
- Tris-HCl, pH 8.0: 400 mM (4 ml of 1 M stock)
- DTT: 50 mM (0.5 ml of 1 M stock)
- Triton X-100: 1% (v/v) (1 ml of 10% stock)
- Spermidine, pH 7.0: 20 mM (0.4 ml of 500 mM stock)
- MgCl2: 200 mM (2 ml of 1 M stock)
- Instructions:
- Add water to make up to 10 ml.
- Components (for 10 ml):
7. Sodium Phosphate Buffer
Sodium Phosphate Buffer is used to maintain a consistent pH during hybridization.
- Preparation:
- Components:
- NaH2PO4: 0.2 M (473.5 ml)
- Na2HPO4: 0.2 M (26.5 ml)
- Instructions:
- Add water to make up to 1 liter.
- Components:
8. Denhardt’s Solution
Denhardt’s Solution is used as a blocking agent during hybridization to prevent non-specific binding.
- 100× Denhardt’s Solution Preparation:
- Components (for 500 ml):
- Ficoll 400: 0.02 g/ml (10 g)
- Polyvinylpyrrolidone: 0.02 g/ml (10 g)
- Bovine Serum Albumin: 0.02 g/ml (10 g)
- Instructions:
- Add water to make up to 500 ml.
- Components (for 500 ml):
9. Hybridization Buffer
Hybridization Buffer facilitates the binding of the probe to the target RNA on the membrane.
- Preparation:
- Components (for 250 ml):
- Formamide: 50% (125 ml)
- SSC: 3× (37.5 ml of 20× stock)
- Denhardt’s Solution: 10× (25 ml of 100× stock)
- Sodium Phosphate Buffer, pH 8.0: 10 mM (25 ml of 100 mM stock)
- EDTA: 2 mM (1 ml of 500 mM stock)
- SDS: 0.1% (2.5 ml of 10% stock)
- Salmon Sperm DNA: 200 μg/ml (10 ml of 5 mg/ml stock)
- Sodium Heparin: 400 U/ml (20 ml of 5000 U/ml stock)
- Instructions:
- Add water to make up to 500 ml.
- Components (for 250 ml):
10. Wash Buffers
Wash Buffers are used to remove non-specifically bound probes during the washing steps.
- Low Stringency Wash Buffer Preparation:
- Components:
- SSC: 2× (100 ml of 20× stock)
- SDS: 0.1% (10 ml of 10% stock)
- Instructions:
- Add water to make up to 1 liter.
- Components:
- High Stringency Wash Buffer Preparation:
- Components:
- SSC: 0.1× (50 ml of 20× stock)
- SDS: 0.1% (10 ml of 10% stock)
- Instructions:
- Add water to make up to 1 liter.
- Components:
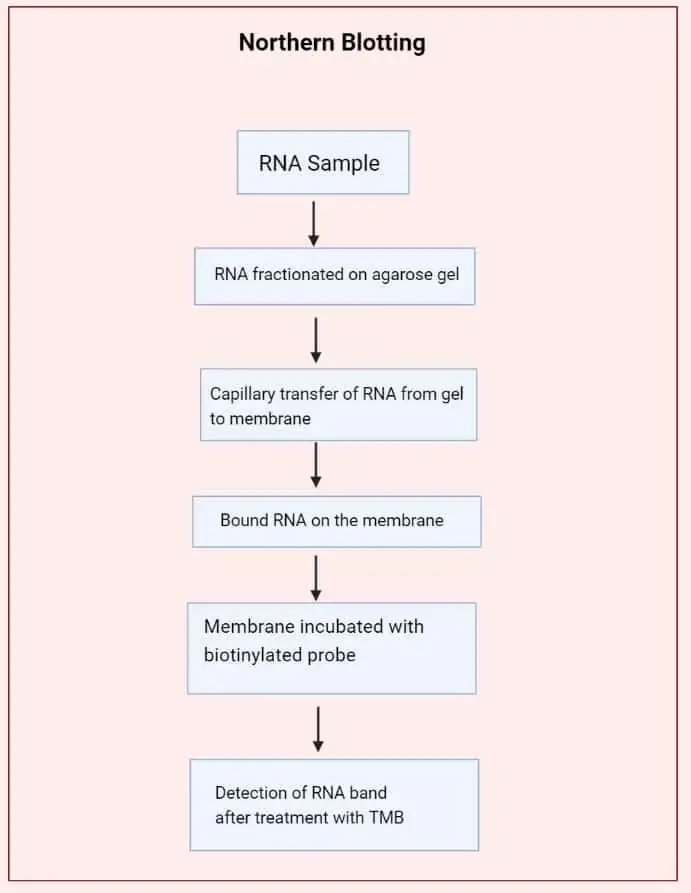
Northern Blotting Protocol
A. Step 1 – Preparation
Proper preparation is crucial for the success of Northern blotting, a technique used to detect specific RNA sequences within a sample. This involves meticulous planning and execution to ensure the integrity of RNA and the accuracy of results. Below is a detailed guide on the preparatory steps necessary for Northern blotting, emphasizing the importance of RNase-free conditions, buffer preparation, and probe generation.
1. Buffer Preparation
Buffers play a vital role in maintaining the appropriate conditions for RNA extraction, electrophoresis, and hybridization. To ensure their effectiveness, it is essential to prepare and sterilize them correctly.
- Steps for Buffer Preparation:
- Formulation: Prepare all required buffers following precise recipes. Each buffer should be mixed thoroughly to ensure all components are dissolved.
- Sterilization:
- Autoclaving: Autoclave the buffers to eliminate any contaminants, particularly RNases, which can degrade RNA.
- Filter Sterilization: For heat-sensitive solutions, use filter sterilization. Pass the buffer through a 0.22-micron filter to remove any potential contaminants.
- Storage: Store the sterilized buffers in RNase-free containers to prevent contamination. Clearly label each container with the buffer name, concentration, and preparation date.
2. Maintaining RNase-Free Conditions
RNA is highly susceptible to degradation by RNases, enzymes that break down RNA. Therefore, maintaining an RNase-free environment is critical throughout the preparation and execution of Northern blotting.
- RNase-Free Techniques:
- Wear Disposable Gloves: Always wear disposable gloves when handling RNA, reagents, and equipment. Change gloves frequently to avoid contamination.
- Use RNase-Free Reagents and Consumables: Ensure all reagents are RNase-free. Use disposable, RNase-free pipette tips and tubes.
- Work in a Clean Area: Designate a specific workspace for RNA work. Clean the area thoroughly with RNase-decontaminating solutions before starting the preparation.
- Proper Storage: Store RNA samples and reagents in RNase-free conditions, typically at -80°C for long-term storage or -20°C for short-term use.
3. Probe Generation
Probes are essential for detecting specific RNA sequences. They are typically generated through PCR (Polymerase Chain Reaction) and must be complementary to the target sequence. If an RNA probe is needed, the template should include a T7 promoter sequence.
- Steps for Probe Generation:
- Designing the Template: Ensure the template DNA includes the sequence of interest and a T7 promoter if generating an RNA probe.
- PCR Amplification: Use PCR to amplify the template. The T7 promoter (TAATACGACTCACTATAGGG) facilitates the transcription of the probe RNA.
- Purification: Purify the amplified product to remove any contaminants that might interfere with hybridization.
4. Handling Radioactive Materials
Radioactive probes are often used in Northern blotting for sensitive detection of RNA. Handling radioactive materials requires strict adherence to safety protocols.
- Safety Protocols:
- Consult Radiation Safety Officer: Follow institutional guidelines for ordering, handling, and disposing of radioactive materials. Obtain necessary approvals and training.
- Proper Storage: Store radioactive materials in designated areas, ensuring they are properly shielded and labeled.
- Use Protective Equipment: Wear appropriate protective gear, including lab coats, gloves, and safety glasses. Use radiation shielding where necessary.
- Waste Disposal: Dispose of radioactive waste according to institutional and regulatory guidelines to prevent contamination and exposure.
B. Step 2 – Separate RNA by a denaturing gel
- Objective: Separate RNA based on size using a denaturing agarose gel with formaldehyde.
- Duration: Approximately 5 hours.
Detailed Steps
- Assemble Agarose Gel Rigs: Set up the apparatus for gel electrophoresis.
- Prepare Denaturing RNA Gel Solution:
- Mix the agarose with the denaturing agents and heat the solution to dissolve.
- Add formaldehyde to denature the RNA.
- Pour the RNA Gel in a Hood: Carefully pour the denatured RNA gel solution into the gel rigs. Use caution to avoid bubbles.
- Solidify the Gel: Allow the gel to solidify. This typically takes about 30 minutes.
- Equilibrate Gel with Running Buffer:
- Before running the gel, soak it in running buffer for at least 30 minutes to ensure uniform conditions throughout the gel.
- Prepare RNA Samples:
- Mix 15 μg of RNA sample with an equal volume of 2× RNA loading buffer.
- Dilute 3 μg of Millennium RNA Markers in the same volume of 2× RNA loading buffer.
- Incubate the samples at 65°C in a heating block for 12-15 minutes, then immediately place them on ice.
- Load Samples onto the Gel:
- Load all samples onto the equilibrated gel, leaving space between the first sample and the RNA marker.
- Run the Gel:
- Run the gel at 125 V for approximately 3 hours. The RNA molecules will migrate through the gel based on their size, with smaller molecules moving faster than larger ones.
Tips and Considerations
- Ensure that the total volume of the sample does not exceed the well volume of the gel comb. If the RNA sample is too diluted, salt can precipitate the RNA. In such cases, resuspend the RNA with less water to prevent this.
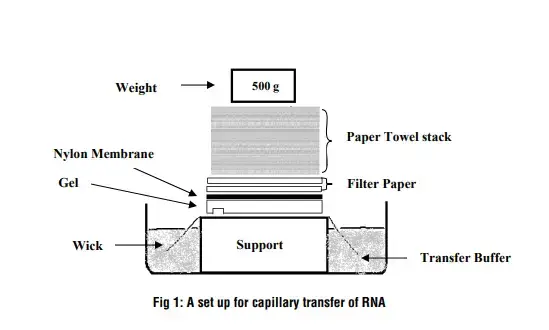
C. Step 3 – Transfer RNA from gel to nylon membrane
- Objective: Transfer RNA from gel to a nylon membrane.
- Duration: Approximately 2 hours.
Detailed Steps
- Prepare Membrane and Filter Paper:
- Cut a nylon membrane to the size of the RNA gel or slightly larger.
- Cut a filter paper to the same size as the nylon membrane.
- Prepare Gel:
- Rinse the RNA gel with water.
- Fill the wells of the RNA gel with melted agarose.
- Prepare Membrane and Filter Paper:
- Wet the nylon membrane and filter paper in water and then in 10× SSC buffer.
- Assembly:
- Place the wet filter paper on the vacuum porous stage.
- Ensure the filter paper is positioned where the cut window of the green plastic gasket will be.
- Place the wetted nylon membrane on top of the filter paper, ensuring no air bubbles are trapped between them.
- Sealing:
- Wet the seal o-ring on the base unit with water.
- Place the plastic gasket on top of the membrane/filter paper, ensuring it covers the seal o-ring.
- Gently place the gel on top of the gasket with the well-side up, ensuring it overlaps with the gasket by at least 5 mm.
- Remove any air bubbles between the gel and the nylon membrane.
- Vacuum Transfer:
- Place the sealing frame on top of the vacuum stage and lock it.
- Start the vacuum source and adjust the pressure to 5 inches of Hg.
- Gently press the gel along the window to apply extra pressure.
- Pour 1 L of 10× SSC buffer into the reservoir and transfer for 90 minutes at 5 inches of Hg. Ensure the buffer level is above the gel.
- Post-Transfer Steps:
- After transfer, remove the sealing frame and drain the buffer.
- Remove the gel and take out the nylon membrane. Dry the membrane between two sheets of filter paper.
- UV crosslink the membrane twice to fix the RNA on the membrane. Use a fine marker to mark the edge of the side with RNA.
- Clean Up:
- Thoroughly clean the gel transfer system by rinsing with plenty of water.
Tips and Considerations
- After transfer, the gel area inside the window of the green gasket should be half as thick as the gel outside the window.
- To check for any remaining RNA, the gel can be illuminated with UV light.
D. Step 4 – Hybridization
- Objective: Use in vitro T7 transcription to make radioactively labeled RNA probes complementary to RNA transcripts of interest and then hybridize them to a membrane.
- Duration: Approximately 18 hours.
Detailed Steps
- Prepare Membrane:
- Place the cross-linked nylon membrane in a hybridization bottle with the RNA-side up.
- Pre-hybridization:
- Add 10 ml of hybridization buffer to the membrane in the bottle.
- Incubate at 68°C for 1 hour to pre-hybridize the membrane.
- Probe Transcription:
- During pre-hybridization, start the probe transcription reaction.
- For a gene probe transcription reaction:
- Combine 10–50 pmoles of DNA template, transcription buffer, ATP, GTP, CTP, [α-P32]-UTP, and T7 polymerase in a PCR tube.
- For a Millennium Marker probe transcription reaction:
- Combine 1 μg of DNA template, transcription buffer, ATP, GTP, CTP, UTP, [α-P32]-UTP, and T7 polymerase in a PCR tube.
- Incubate at 37°C for 2 hours.
- Add DNase I and incubate at 37°C for 15 minutes to digest the template.
- Probe Purification:
- Use a micro G-50 sephadex spin column to purify the probe.
- Load the transcription reaction onto the column and spin.
- Add the purified probe to scintillation liquid and check the counts.
- Hybridization:
- Dump the pre-hybridization solution and add hybridization buffer with probes to the hybridization bottle.
- Hybridize overnight at 68°C.
- Washing:
- Pre-warm low stringency and high stringency buffers at 68°C.
- Wash with low stringency buffer for 5 minutes twice.
- Wash with high stringency buffer for 15 minutes twice.
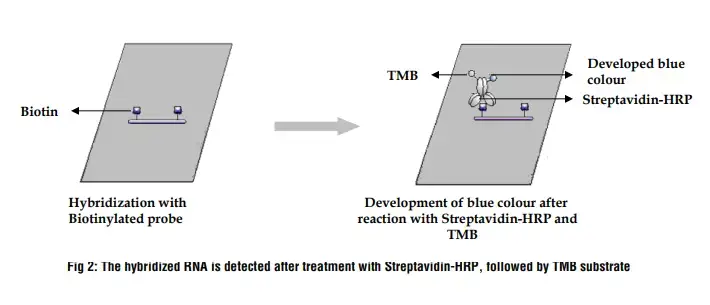
- Detection:
- Take out the membrane and briefly dry it on a kimwipe.
- Wrap the membrane with saran wrap and expose it to a phosphor screen overnight for detection.
Tips and Considerations
- If the gene probe is longer than 400 nt, increase the limiting nucleotide concentration (UTP) in the transcription reaction to 5–25 μM by adding more non-labeled UTP.
Observation and Result
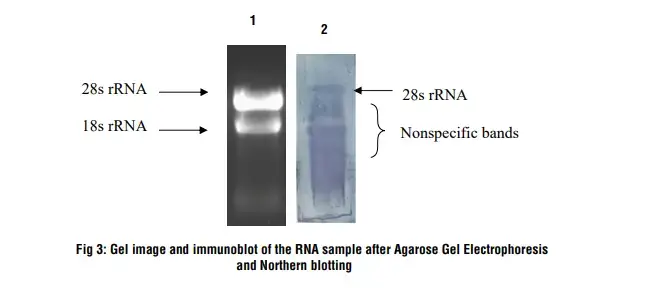
- Lane 1: RNA sample on 1.2 % denaturing agarose gel
- Lane 2: After Northern hybridization a blue band develops on the nylon membrane
Precautions
- Prior to start the experiment, the electrophoresis tank should be cleaned with detergent solution (e.g., 0.5% SDS), thoroughly rinsed with RNase-free water, and then rinsed with ethanol and allowed to dry.
- Tips, pipettes, electrophoresis unit etc to be used for the experiment must be UV treated for 15-20 minutes.
- Use sterile, disposable plasticwares and micropipettes reserved for RNA work to prevent cross-contamination with RNases from shared equipments.
- Use RNase-free water for diluting the solutions.
Northern blotting vs Southern blotting
| Northern Blotting | Southern Blotting | |
|---|---|---|
| Target Molecules | RNA molecules | DNA molecules |
| Purpose | Detection and analysis of RNA molecules | Detection and analysis of DNA molecules |
| Technique | Gel electrophoresis, membrane transfer, hybridization | Gel electrophoresis, membrane transfer, hybridization |
| Target Sequence | Complementary DNA (cDNA) or RNA probes | DNA probes |
| Applications | Gene expression analysis, RNA processing studies, non-coding RNA analysis | DNA analysis, gene mapping, detection of specific DNA sequences |
| Detection Method | Radioactive or non-radioactive labeling of probes | Radioactive or non-radioactive labeling of probes |
| Sensitivity | Generally lower sensitivity compared to newer RNA analysis methods such as qRT-PCR or RNA-seq | Can detect low-abundance DNA sequences, high sensitivity possible with appropriate probes and conditions |
| Sample Requirements | Requires a relatively large amount of RNA sample | Requires a relatively large amount of DNA sample |
| Limitations | Time-consuming, lower sensitivity, limited dynamic range for quantification | Time-consuming, potential for DNA degradation, limited dynamic range for quantification |
| Key Applications | Gene expression analysis, mRNA splicing analysis, non-coding RNA analysis | DNA mapping, detection of specific DNA sequences, analysis of DNA mutations or polymorphisms |
Application of northern blotting
- Gene Expression Analysis: Northern blotting is commonly used to study gene expression patterns in different tissues, developmental stages, or disease conditions. By analyzing the abundance of specific mRNA molecules, researchers can determine the level of gene expression and compare it between different samples. This application has contributed to our understanding of gene regulation, cellular processes, and the identification of differentially expressed genes.
- mRNA Splicing Analysis: Northern blotting can provide insights into mRNA splicing patterns. By probing for specific splice variants of a gene, researchers can determine the presence and abundance of alternative mRNA isoforms. This helps in understanding the regulation of alternative splicing events and their functional implications.
- RNA Processing and Stability: Northern blotting allows the investigation of RNA processing events, such as RNA cleavage, polyadenylation, and stability. By examining the size and abundance of specific RNA fragments, researchers can assess the processing efficiency and stability of RNA molecules. This information is crucial for understanding RNA metabolism and post-transcriptional regulation.
- Non-coding RNA Analysis: Northern blotting has been instrumental in the identification and characterization of non-coding RNAs, such as microRNAs (miRNAs) and long non-coding RNAs (lncRNAs). These non-coding RNAs play important roles in gene regulation, development, and disease. By designing specific probes, researchers can detect and quantify these non-coding RNAs, providing insights into their expression patterns and potential functions.
- Validation of Transcriptomic Data: Northern blotting can serve as a valuable technique for validating transcriptomic data obtained from high-throughput methods, such as RNA sequencing (RNA-seq). It allows the confirmation of gene expression changes observed in large-scale datasets by directly assessing the expression of specific mRNA molecules.
- Diagnostic Applications: Northern blotting has been used in clinical settings for the diagnosis of certain diseases. By detecting the presence and abundance of disease-related RNA molecules or RNA biomarkers, researchers can assess disease status or monitor treatment responses.
- Comparative Analysis: Northern blotting can be utilized to compare RNA expression levels between different samples, such as healthy and diseased tissues, control and experimental conditions, or different individuals. This comparative analysis helps identify molecular differences associated with specific conditions or treatments.
Northern Blotting Advantage
- Detection of Specific RNA Molecules: Northern blotting allows the specific detection and analysis of RNA molecules of interest. By using complementary probes, researchers can target and detect specific RNA sequences, providing insights into gene expression, splicing patterns, and non-coding RNA expression.
- Size Determination: Northern blotting provides information about the size of RNA molecules. By comparing the migration of RNA samples with size markers on the gel, researchers can determine the approximate size of the target RNA molecules, which can be important for understanding RNA processing and transcript variants.
- Validation of Gene Expression Data: Northern blotting can serve as an independent method to validate gene expression data obtained from other techniques, such as RNA-seq. It allows for the confirmation of gene expression changes observed in large-scale datasets by directly assessing the expression of specific mRNA molecules.
- Cost-effective: Compared to some high-throughput techniques like RNA-seq, Northern blotting is a relatively cost-effective method. It does not require expensive equipment or reagents, making it accessible for laboratories with limited resources.
- Customizability: Northern blotting allows researchers to design and generate specific probes for their target RNA molecules. This flexibility enables customization and adaptability to specific research questions or RNA targets of interest.
Northern Blotting Disadvantage
- Time-consuming and Labor-intensive: Northern blotting is a time-consuming technique that requires multiple steps, including RNA extraction, gel electrophoresis, transfer, hybridization, and detection. It involves several manual manipulations and can take several days to complete, making it labor-intensive.
- Low Sensitivity: Compared to newer techniques like quantitative real-time PCR (qRT-PCR) or RNA-seq, Northern blotting may have lower sensitivity. The detection limit of Northern blotting depends on the abundance of the target RNA and the efficiency of probe hybridization. It may not be suitable for detecting low-abundance or rare RNA molecules.
- Limited Dynamic Range: Northern blotting may have a limited dynamic range for quantifying RNA expression. The intensity of the hybridization signal on the blot does not always correspond linearly to the abundance of RNA in the sample. It may be challenging to accurately quantify RNA expression levels using Northern blotting.
- Sample Degradation: RNA is prone to degradation by RNases, making it necessary to handle RNA samples with care. Contamination or improper handling can lead to RNA degradation, affecting the quality and integrity of the RNA samples and potentially compromising the results of Northern blotting.
- Relatively Large Sample Requirements: Northern blotting typically requires a relatively large amount of RNA sample, making it challenging when working with limited or precious sample materials. Extraction of sufficient RNA can be difficult, particularly for small or rare cell populations.
FAQ
What is Northern blotting?
Northern blotting is a molecular biology technique used to detect and analyze RNA molecules. It involves the separation of RNA fragments by gel electrophoresis, transfer to a solid membrane, and subsequent hybridization with specific probes to visualize and study RNA molecules of interest.
What is the purpose of Northern blotting?
The primary purpose of Northern blotting is to investigate gene expression patterns, analyze mRNA abundance, study alternative splicing events, and detect specific RNA molecules in biological samples.
How does Northern blotting differ from Southern blotting?
Northern blotting focuses on the detection and analysis of RNA molecules, whereas Southern blotting is used for DNA analysis. While both techniques involve gel electrophoresis and probe hybridization, they differ in terms of the target molecules and their detection.
What are the key steps involved in Northern blotting?
The main steps in Northern blotting include RNA extraction, electrophoresis, transfer to a membrane, hybridization with specific probes, and detection of the RNA molecules. These steps allow for the separation, immobilization, and visualization of RNA molecules.
How are the RNA molecules immobilized on the membrane during Northern blotting?
After gel electrophoresis, the separated RNA fragments are transferred from the gel to a solid membrane, typically through a capillary or vacuum-based method. The RNA molecules adhere to the membrane, where they can be probed and detected.
What types of probes are used in Northern blotting?
Probes used in Northern blotting are typically complementary DNA (cDNA) or RNA molecules that are labeled with radioactive or non-radioactive tags. These probes hybridize with the target RNA sequences, allowing their detection.
Can Northern blotting detect different types of RNA molecules?
How is the specificity of probe hybridization ensured in Northern blotting?
The specificity of probe hybridization is ensured by designing probes that are complementary to the target RNA sequences. Stringent hybridization conditions, including temperature and buffer composition, are optimized to maximize the specificity of probe binding.
What are the limitations of Northern blotting?
Some limitations of Northern blotting include its time-consuming nature, lower sensitivity compared to newer techniques, limited dynamic range for quantification, sample degradation risks, and relatively large sample requirements.
What are the alternative techniques to Northern blotting for RNA analysis?
Alternative techniques for RNA analysis include quantitative real-time PCR (qRT-PCR), RNA sequencing (RNA-seq), microarray analysis, and in situ hybridization. These techniques offer higher throughput, greater sensitivity, and more comprehensive analysis of RNA molecules compared to Northern blotting.
References
- He SL, Green R. Northern blotting. Methods Enzymol. 2013;530:75-87. doi: 10.1016/B978-0-12-420037-1.00003-8. PMID: 24034315; PMCID: PMC4287216.
- https://en.wikipedia.org/wiki/Northern_blot
- https://himedialabs.com/TD/HTBM028.pdf
- https://www.thesciencenotes.com/northern-blotting-principle-procedure-and-applications/
- https://www.mybiosource.com/learn/testing-procedures/northern-blotting/
Related Posts
- Western Blot – Protocol, Principle, Result.
- Polymerase Chain Reaction (PCR) – Definition, Steps, Principle, Application
- Blood Grouping Test – Principle, Procedure, Result
- Gel Staining Procedure for PAGE
- Laboratory Rules – Basic Rules Of A Microbiology Laboratory